Steps for Jar Testing Scale Inhibitors in Oil, Gas Applications
by David Wilson and Kelly Harris
There are two primary sources of water that are a concern during oil production. The first is "formation water" that comes from the oil reservoir itself. The second is water that returns to the surface from intentional water floods carried out to maintain reservoir pressure and oil production flow rates. Invariably these waters are at elevated temperatures as a result of their depth below the earth's surface.
As the water comes to the surface in co-production of oil, the dissolved carbon dioxide comes out of solution as pressure is reduced. This leads to a pH increase and an equilibrium change where the bicarbonate anion decomposes to the carbonate anion and more carbon dioxide. The formation of the carbonate anion and pH increase gives rise to calcium carbonate scaling that can reduce the internal diameter of pipes and eventually block pipes and pumps used in separation facilities.
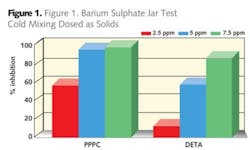
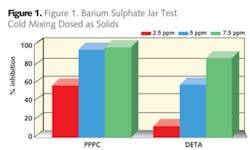
In the case where water floods are performed it is unusual to have the desired quality of injection water and invariably incompatibility between the injection water and the formation water occurs. This can be manifested by an increase in carbonate scaling tendency or by the production of other scales such as calcium, barium or strontium sulphates. These scale types are difficult to remove by dissolution techniques so it is prudent to apply scale inhibitors to help prevent their deposition.
However the scale inhibitors themselves can cause problems. Sodium hexametaphosphate and other polyphosphates have been used for controlling calcium carbonate or barium sulphate deposition in oil wells. Polyphosphates precipitate in the presence of high calcium levels and are susceptible to hydrolysis and reversion back to the orthophosphate ion. This in turn can lead to calcium phosphate deposition.
Scale inhibitors commonly used at present consist of phosphate esters, phosphonates, polyacrylates, phosphinopolyacrylates, polymaleic acids, terpolymaleic acids, sulphonic acid copolymers, polyvinyl sulphonates, and more recently the so called 'green' inhibitors polyaspartaic acid and carboxy methyl inulins.
The mechanisms by which inhibitors work is varied and each inhibitor, to a greater or lesser extent, have characteristics of all the mechanisms. These mechanisms are generally accepted to be:
- Threshold.
- Growth inhibition / crystal distortion
- Dispersancy.
Threshold inhibition is where the inhibitor is involved at the early equilibrium stages of crystal formation disrupting ion clusters before they reach critical size for nucleation. As a result the ions dissociate, releasing the inhibitor to repeat the process, hence the low dose levels required. To do this the inhibitor must bind scale forming ions, but unlike with chelants the bound ions must be available to interact with their counter ions.
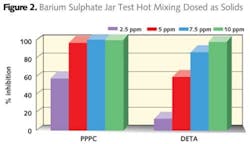
Simplistically, fast crystal growth leads to edges and points. Inhibition of these active sites by lattice matching of the scale inhibitor to the crystal lattice will slow the growth of the crystal, the inhibitor being consumed by incorporation into the lattice. Usually this leads to a distorted crystal lattice which is usually softer than uninhibited crystal exhibiting a dust like appearance on surfaces. These crystals typically are not as adherent as the uninhibited crystals.
For dispersancy to take place, just as in growth blocking, the inhibitor must bind to the surface. To be an effective dispersant though, it has to present a charged group to the solution such that other charged particles are repulsed.
Inhibitor deployment in oilfields needs to balance many considerations regarding the proposed inhibitors. The list below covers some of these issues:
- Minimum inhibitor concentration (MIC) required to prevent scaling.
- Compatibility of inhibitor with brines and formation waters.
- Compatibility with other chemicals used in the system.
- Hydrothermal stability.
- Adsorption and desorption to and from the reservoir rock.
In addressing the MIC considerations, the tests that have been developed to identify and select inhibitors for use are jar testing or threshold test and dynamic scale loop tests. The classic jar test appears to be a simple test, only requiring the mixing of appropriate waters in the presence of inhibitor and heating at temperature for a specified timeframe.
However, there are issues to consider: What type of jar does one use? That generally used for powders or the Winchester type? What type of insert is required, plastic, card, or rubber septum? Will it matter?
In the case of calcium carbonate testing, yes, it will matter. Jar types are made within quite large tolerance which means that their size can vary considerably (typically in 120ml jars ± 5 to 10ml). If a standard 100ml is used in each test then the head space above this 100ml can vary. The volume of the headspace then dictates the severity of the test as carbon dioxide is released into it. The more carbon dioxide being released the more severe the test.
One could say that this is being somewhat pedantic, but coupled with the kind of insert used in the bottle this becomes significant. Card inserts allows carbon dioxide to escape to atmosphere thus allowing the bicarbonate decomposition reaction to continue and drive the severity of the test. Plastic and rubber inserts are generally more reliable and acceptable. In the case of barium or calcium sulphate jar tests there is little impact of headspace or cap issues, since pH does not drive their precipitation.
Having overcome the choice of jars and cap inserts the next stage is to decide how to make the waters. Should the waters be made as formation water and aquifer water / seawater separately or as separate anion and cation solutions? How to achieve the pH of the test, saturation with carbon dioxide, use acid or with buffers? These decisions are usually based on the water analysis from the field under consideration. Experience has taught that making formation water and aquifer water / seawater as separate mixes is fraught with problems.
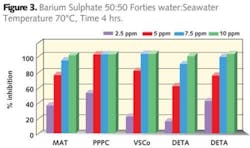
It is more prudent to make separate solutions, one containing scaling cations and the other containing the scaling anions. The ionic strength of these two solutions being made as close as possible, balanced with sodium chloride. To achieve the requisite pH both solutions should be buffered. The use of carbon dioxide to saturate the waters should be avoided. It is impractical when considering the points made in jar and cap insert selection. The use of acids to buffer the solutions should also be avoided. Addition of an acid to a pre-determined pH, for example pH 5.5, is easily obtained by a few drops of acid. This does not mean that the pH will remain at this level throughout the test. It will invariably increase as this method will not destroy the bicarbonate present. In order to ensure that the pH remains at pH 5.5 the solutions require air blowing with the addition of the acid to a permanent pH 5.5.
The selection of acid is also important. The use of sulfuric acid should be avoided as this may then give rise to calcium sulphate scaling in water that would not otherwise give this scale. By far the most appropriate way of obtaining the pH required is by the use of buffers, acetic acid and sodium acetate being the best.
The next consideration is order of addition of the waters and inhibitors to the jars. This is of paramount importance. The inhibitor should not be mixed with the cation solution as the inhibitor may have calcium intolerance and would precipitate prior to the full waters being mixed. This would result in the inhibitor not being at the design level and would also give rise to a nucleation site for scale to form. Order of waters placed in the jar should therefore be inhibitor or anion solution first or second and the cation solution added last with good mixing.
Should the test waters be pre-heated and should a water bath or oven be used for heating the solutions?
For calcium carbonate testing it is better if the solutions are not pre-heated due to the inverse solubility of calcium carbonate. However in the case of barium sulphate the solutions should be pre-heated. Hot mixing gives improved results for the phosphonate (DETA) and much lower dose levels are required for both the phosphinocarboxylic acid (PPCA) and DETA. These observed differences are actually the impact of temperature on the saturation index (SI) of the barium sulphate, the lower temperature giving a more severe condition and higher SI than that of the hot mixed waters.
Should a water bath or oven be used as heating source?
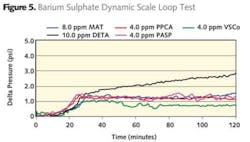
Ovens suffer from the same issue as cold mixing. Once opened to allow placement of jars the temperature drops and some time is taken to achieve the desired test temperature. So again a more severe condition exists. In the case of water baths the heat transfer is much better although the temperature is limited to below 100°C. Figure 3 demonstrates the performance of a range of additives evaluated using hot mixing of test waters. Comparison of a maleic acid terpolymer (MAT) with PPCA, a sulphonated copolymer (VSCo), DETA and a polyaspartate (PASP) demonstrate that all inhibit barium sulphate.
Should the test be homogeneous or heterogeneous?
If homogeneous, then the mechanism tested is that of threshold. If heterogeneous, by addition of seed crystal or sand, then growth inhibition is been tested simultaneously with threshold.
How long should the duration of the test be?
It should cover the residence time in the system to the point where the water has been separated from the oil and is ready to be discharged or re-injected. So the time needs to be four hours minimum. Longer time periods give a complexity to the results as once scale starts to form the mechanism of the test changes from that of a homogeneous test to one of a heterogeneous test.
A simple jar test? Not really!
For the dynamic scale loop the same considerations are required as for the jar test with regards to making the water, as well as heating and mixing it. In addition other parameters need to be taken into account, such as length and diameter of tube, tube metallurgy, flow regime (velocity through the tube), pump type and calibration of pump and whether to pre-scale the tube or start from a clean condition.
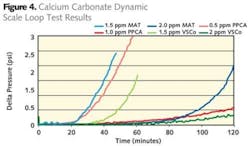
In general it is accepted that the tube is made of stainless steel 1m long and 1mm in diameter. Flow rates used are typically in the 0.5m/s region. This means that the residence time in the coil is approximately 2 seconds. Tests are usually conducted over a 2 hour period with a precondition of less than 1 psi pressure rise over this time frame. Figure 4 shows comparisons of MAT, PPCA, VSCo under calcium carbonate scaling conditions with no pre-scaling and Figure 5 shows comparisons of MAT, PPCA, VSCo, DETA and PASP for barium sulphate using a pre-scaled system. As can be seen from the data all the additives perform well.
However differentiation can be seen in the dose levels used to meet the criteria specified earlier. Where very high levels of barium and sulphate are expected to comingle in the reservoir, laboratory simulation invariably blocks the tube before any inhibitor can reach the pre-scaled system. In these cases it is better to start with a clean tube.
So now we have good tools for evaluating inhibitors in both jar and dynamic conditions and can separate out good from poor performers and also determine the MIC level for scale control. This is not the end of the story though as not only must the inhibitors perform but they must be compatible with the brine in the field, especially when applying Squeeze treatments.
Tests have been designed to identify the phase envelop of inhibitors under varying brine, pH, inhibitor concentrations, and temperatures. The reason for this is twofold:
- Avoid precipitation in the near well bore area
- Identify potential candidates for phase separation Squeezing.
Other compatibility studies are also needed to determine if the inhibitor goes through a calcium incompatibility phase during the return to MIC after a Squeeze treatment. Such behavior may preclude use of the additive as this calcium inhibitor complex could act as a scale or be a seed for further scale deposition.
Hydrothermal stability
Hydrothermal stability testing is a requirement for additives as they are in contact with the reservoir for long time periods. Typically these tests are performed in small Teflon lined autoclaves. The diluted inhibitor in seawater is nitrogen sparged to remove oxygen and then subjected to the reservoir temperature in question for a specific time frame. After this treatment the inhibitor solution is inspected for obvious physical changes, analyzed for chemical changes and finally subjected to performance testing to determine any loss in activity.
These tests may be flawed in their approach as due to the free rotation of bonds, the solution phase inhibitor is more susceptible to hydrolysis / cleavage, than the inhibitor that is adsorbed onto a rock surface. Some testing performed by RMS Wat et al, in Statoil, based on a core flood technology has demonstrated that an additive has better hydrolytic stability than the equivalent test in an autoclave. The implication of this is that perfectly adequate additives may have been discarded because the test designed for selection is flawed.
Unless this issue is addressed things will only get worse with the advent of HPHT wells putting increasingly higher temperature and pressure demands on the inhibitors. Longer subsea tiebacks and pipelines onto shore base separation facilities also means that oil water mixtures will be at seabed temperatures of approximately 4°C (39.2°F) with the possibility of gas hydrate, naphthenate and asphaltene formation.
The time is ripe to see if the present tests can meet the demands of the oil industry. Are the jar and dynamic scale loop test fit for purpose?
For the HPHT conditions, the jar test needs modification such that it can be performed in a small autoclave. This in itself poses significant issues as hot mixing at reservoir temperature is impractical. An alternative is to design a different water to the particular formation / seawater mix by mimicking the saturation index of this water at a lower temperature than is achievable with the present test. Yes, this has drawbacks but it may be the best compromise. The data generated under these conditions may then be checked by evaluating the inhibitor in the HPHT dynamic loop tests where pre-heat coils are used to heat the anions and cations prior to their mixing in a T-piece before the test coil.
Addressing the seabed temperature issues in some ways are easier as the existing jar and dynamic scale tests will be adequate. Again modifications may be necessary to incorporate other inhibitors in the waters, for example eg methanol which is used for gas hydrate inhibition. Methanol in water will tend to decrease the solubility of most scales and make the true saturation index higher than the calculated one. Inhibitors that have been evaluated in water only mixes will perform less well in the field due to this difference in solubility.
The colder conditions and longer residence times in pipelines will demand higher levels of some inhibitors, particularly for gas hydrate formation and barium sulphate scale. This means that more attention to inhibitor / inhibitor reactions and their compatibility is required. The potential interactions could negate any activity that the inhibitor possesses alone.
More recently there have been scales that are called exotic scale such as iron, lead and zinc sulphide forming in the wells. The present tests do not encompass these types of scales and in the case of iron sulphide presents a particular issue as exclusion of oxygen to prevent oxidation of iron (II) to iron (III) is required. Some institutes are investigating these scales but testing is in the early stages. It is likely that the existing inhibitors will inhibit these 'new' scales to a greater or lesser extent.
About the Authors: Kelly Harris has a PhD in Synthetic Organic Chemistry at The University of Bath. Since BWA Water Additives in 2006, she has developed a number of scale inhibitors for oilfield and industrial water applications. She also has contributed to the development of oilfield and industrial water applications research. David Wilson joined BWA Additives 40 plus years ago when it was Geigy, starting as a Synthetic Organic Chemist. He joined the Water Treatment Applications Laboratory in 1981 developing new products and test methods for oilfield and industrial water treatment applications. Now in the role of Technology Manager, he investigates new products for mitigating scale and corrosion issues in industry.
Past IWW Issues